
プラセボ対照試験とは|GCPレター第68号
■この記事は、2021年1月31日に発行されたGCPレターを転記したものです。
■関連資料
→GCPレター第68号(2021年1月発行)プラセボ対照試験
■GCPレターとは:
臨床試験全体の質の向上とGCPの啓発活動の一環として、当社提携医療機関向けのGCP関連などのレターを2021年3月まで発行いたしました。
一般に、 新薬候補の有効性を検証する場合、プラセボと比較することが原則と考えられています。プラセボを用いた臨床試験をプラセボ対照試験と呼び、これは 新薬候補の有効性を科学的に明らかにするために必要な試験です。
しかしながら、重篤な疾患であって、標準的治療法が確立している場合等においては、プラセボを対照として比較試験を行うことは、倫理的に困難であると言われています。今回は、「プラセボ対照試験」 について見てゆきましょう。
目次[非表示]
プラセボ(placebo)とは
プラセボは、本物の薬とそっくりに見えるよう作られていますが、薬効成分を含まないもので、偽薬とも呼ばれます。
プラセボには薬効成分は入っていませんが、『薬を飲んだ』 と思うだけで心理的作用が働き、症状の改善や副作用の出現が見られることがあります。この現象を、『プラセボ効果』 と言います。
プラセボ対照試験の必要性
臨床試験においてプラセボ効果が働いてしまうと、正しい試験結果を出すことができません。そのため、プラセボ効果を除外し、 客観的な評価を行うために、『プラセボ対照試験』が行われています。
プラセボ対照試験の必要性を示した臨床試験の一つとして、1980年代に行われた、プラセボ対照二重盲検ランダム化比較試験『CAST試験(Cardiac Arrhythmia Suppression Trial)』 があげられます。
CAST試験とは、心筋梗塞患者のうち心室性不整脈の出現が少ない患者のほうが予後が良いこと、また、心筋梗塞で不整脈を持つ患者に抗不整脈薬を投与すると不整脈が減少することから、抗不整脈薬治療により、心筋梗塞患者の死亡率が減少するであろうという仮説を設定、検証した臨床試験です。
CAST試験において、プラセボ投与群に比べて実薬投与群(当時最も強力で新しい抗不整脈薬であったフレカイニドやエンカイニドなどのIc群抗不整脈薬を投与された群)では生存率が減少、言い換えると、実薬を投与することにより2.5倍ぐらい死亡率を高めたという結果が得られました(下図)。
当時、心筋梗塞患者に対する抗不整脈薬治療はすでに行われていました。CAST試験により、不整脈を持つ心筋梗塞患者に対する治療方針は大きく変わり、Ic群抗不整脈薬の投与が激減し、結果的に多くの患者の死を防いだと考えられています。
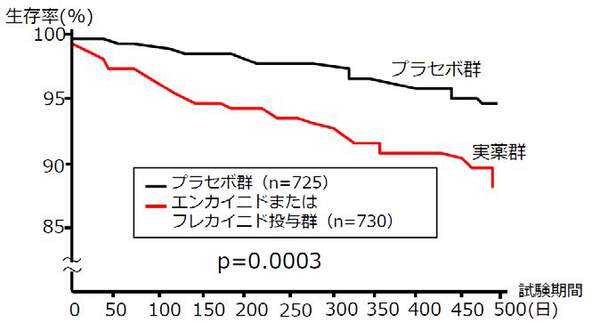
(引用)The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. Preliminary report : effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989; 321(6): 406-12.
CAST試験によって、 新薬候補の有効性は、実際に臨床試験を実施してみないと分からないということ、倫理性が確保された状況下で科学的・客観的な評価が必要であること、さらに、科学的・客観的評価のために、プラセボ対照試験が必要であることが示されました。プラセボ対照試験は、臨床試験に関わるバイアスを最小化し、より信頼性のある結果を得るために生み出された方法です。
自然治癒や生活環境・習慣、併用薬·併用療法といった自然変動のもたらすバイアス、患者が抱く医師への信頼や、 新薬候補に対する期待のバイアスによっても治療効果は生まれます。
プラセボによっても生じる有効性を凌駕する部分がなければ、本当に有効な治療薬とは言えません。
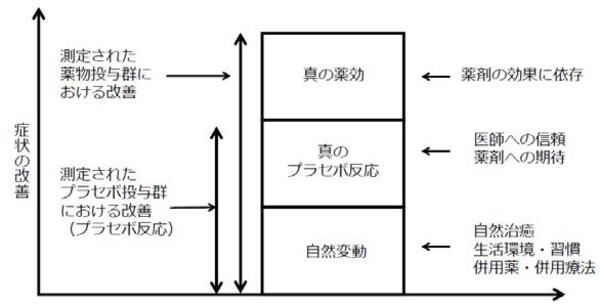
(引用)石田光裕ほか「プラセポ対照試験と被験者公募のポイントー抗うつ薬の開発経験から」 『薬理と治療』 2012;40(2):113-21.
プラセボ対照試験の倫理上の問題
確立された治療法がない場合やリスクが小さい場合には、原則としてプラセボの使用が許容されるというのが一般的な考え方です。しかしながら、既に確立された治療法があるがプラセボを使うという場合には、倫理上の問題が生じます。
研究対象母集団において死亡や回復不能の障害のような重篤な障害を防ぐ治療が利用できることが知られている場合には、 プラセボの使用は一般に不適当です。しかし、重篤な障害が生じない場合には、たとえ患者が結果として不快・不便を経験する可能性があるとしても、プラセボ対照試験への参加を患者に依頼することは一般的に非倫理的ではないと考えられています。
例えば鼻炎に対する新規の抗ヒスタミン剤を開発するというときに、仮に標準治療薬があったとしても、プラセボを使うことが、直ちに非倫理的だとは言えません。
プラセボ単独投与が許容される条件については、ヘルシンキ宣言(2013年)および、CIOMS(Council for International Organizations of Medical Sciences:国際医学団体協議会)による「人間を対象とする健康関連研究の国際的倫理指針」2016年)(以下、CIOMS指針)において、以下のように定められています。
ヘルシンキ宣言(2013)第33項 プラセボの使用
(世界医師会︓ヘルシンキ宣言―人間を対象とする医学研究の倫理原則 (日本医師会訳)、2013(http://dl.med.or.jp/dl-med/wma/helsinki2013j.pdf)一部改変)
- 新しい治療の利益、リスク、負担および有効性は、以下の場合を除き、最善と証明されている治療と比較考量されなければならない:
- 証明された治療が存在しない場合、プラセボの使用または無治療(以下、「プラセボの使用等」)が認められる
- 最善と証明されたものより効果が劣る治療、プラセボの使用等が、その治療の有効性あるいは安全性を決定するために必要な場合
- 最善と証明されたものより効果が劣る治療、プラセボの使用等が、最善と証明された治療を受けなかった結果として、重篤または回復不能な損害の付加的リスクを被ることがないと予想される場合
- この選択肢の乱用を避けるため徹底した配慮がなされなければならない
CIOMS 指針 5︓臨床試験における対照群の選択
(CIOMS指針(栗原千絵子、齊尾 武郎 訳 渡邉 裕司 監修).臨床評価、45(4); 770-774, 2018)
研究対象の疾患に対する効果の確立した介入が存在しない場合には、対照群にプラセボを使うことがあり、また、効果の確立した介入にプラセボが追加されることもある。効果の確立した介入が存在する場合に、効果の確立した介入を提供せずにプラセボを対照群に使用できるのは、以下の両方に該当する場合のみである。
- プラセボを使用する必要不可欠な科学的理由(compelling scientific reason)が存在する。
- 効果の確立した介入を遅らせ、あるいは差し控えることが、参加者に最小限を僅かに超えるリスクの増加(minor increase above minimal risk)しかもたらさず、効果的なリスク低減措置の使用を含めリスクが最小化(risks are minimized)される。
すでに標準治療薬が存在する場合には、その標準治療薬を実薬対照として、同等性試験あるいは非劣性試験を行うべきであると言われています。ただし、非劣性試験を繰り返すと、有効性のないものが世に出てくる可能性も否定できません。これはどういう メカニズムで起こってしまうのかを次の項でご説明します。
非劣性試験とその問題点
通常、臨床試験では、新薬候補が対照(プラセボ、標準治療薬等)よりも有効性があることを検証します(これを優越性試験と言います)。 しかしながら、優越性試験以外にも「効果=ベネフィット」と「安全性=リスク」を秤にかけた臨床試験が行われています。例えば、「新薬候補は標準治療薬と比較して効果は同じだが副作用は少ない」、「新薬候補は標準治療薬と比較して効果も副作用も同じだが、他のメリット(廉価である、投与間隔が長い等)がある」 などが想定される場合が考えられます。
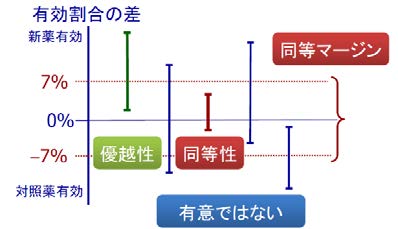
有効性が標準治療薬と同程度であることを示すことにより、新薬候補の有効性を証明する臨床試験を「同等性試験」といいます。同等性試験では、「この範囲に収まっていれば、2つの薬は有効性が同等だと判定してもいい」という範囲を事前に決めます。 これを「同等マージン」と呼んでいます。
下図では、同等マージンを±7%としています。
信頼区間を計算して、試験①のような幅になれば、信頼区間の下限が“0%”よりも上で 新薬有効となり、優越性が示された試験と言えます。
それに対して、信頼区間に“0%”を含んでいる試験(試験②、③、④)あるいは、試験⑤のように完全に”0%“よりも下回ってしまっている試験は優越性は示せません。次に、同等性について見てゆきましょう。
同等性試験の場合は、まず同等性マージンを決め、同等マージンの範囲に収まっていれば、同等性が示されたことになります。この例の場合、試験③は、同等マージン内に入っており、試験③だけが同等性を示せたことになります。
先ほど優越性が示されたと言った試験①についても、同等マージンの範囲をはみ出していますので、試験①②④⑤においては、同等性は証明できなかったことになります。
(引用)第4回 プラセボ対照試験に関する専門部会(2015年5月8日) 資料2、一部改変
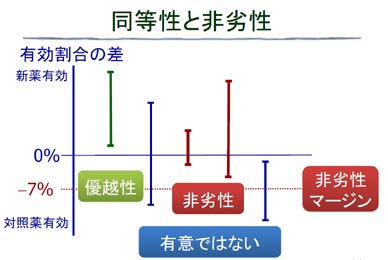
ジェネリック医薬品の場合は、先発医薬品よりも良くても悪くてもいけないので同等性を検証する必要がありますが、それ以外の場合には、標準治療薬よりも有効性が一定以上劣らないことを証明する「非劣性試験」が行われています。
非劣性試験では、マージンは下限だけを設定します。非劣性試験のマージンの考え方としては、標準治療薬よりも一定以上劣 ったら、有効性として許容できない限界であるということが、臨床的な観点から決められます。1つの考え方として、最初に標準治療薬が承認されるときには、おそらくプラセボ対照試験を行って承認されているので、少なくともプラセボとの有効割合の差よりも狭く設定しないと、有効性がプラセボよりも悪いものが承認されてしまう可能性もあります。例えば、「プラセボとの差の2分の1」などを 「非劣性マージン」として決めることになります。
下図では、非劣性マージンを-7%としています。
試験①だけ「優越性」と記載されていますが、これも非劣性試験の範疇から言うと非劣性が証明されたことになります。
また、試験③と試験④も信頼区間の下限が非劣性マージンよりも上にあるので非劣性が 証明できたと考えることができます。
試験②と試験⑤は、信頼区間の下限が非劣性マージンよりも下にあるので非劣性が証明できなかったと考えます。これが非劣性試験の考え方になります。
(引用)第4回 プラセボ対照試験に関する専門部会(2015年5月8日) 資料2、一部改変
非劣性マージンの設定によっては、有効性で明らかに対照薬よりも劣っている新薬が、承認される可能性があります。実例として、2013年に承認されたアクテムラ®皮下注を挙げて説明します。
アクテムラ®皮下注は関節リウマチの抗体薬で、すでに点滴静注製剤が承認されていましたが、皮下注の場合、自宅でできるため、利便性に勝るということで、点滴静注製剤との非劣性試験が実施されました。
最初に点滴静注製剤が承認されたときの試験では、標準治療薬「メトトレキサート」との24週の関節リウマチの改善率の差が57.3%(プラセボとの差(12週)は65.5%)と著効している薬でした。
メトトレキサートとの差の3分の1が19.1%でしたので、この試験での非劣性マージンとして-18%が設定され、試験結果は、信頼区間が-17.6~-1.2%でした。非劣性マージンが-18%ですので、この試験では、非劣性が証明されましたが、信頼区間の上限が“0%”を下回っており、有効性では、明らかに点滴静注製剤よりも劣っている薬だといえます。 このように、非劣性マージンの範囲内で劣った新薬候補が承認されてしまうことは非劣性試験に特有な問題点です。
少し劣った医薬品が次の対照薬となり、また新しい新薬候補と非劣性試験を行う、また、それが少し劣ったものとして、新しい新薬候補と非劣性試験を行う、このように順々につないでいくと、最終的にはプラセボと変わらない医薬品が承認されてしまうことも、原理的には起こる可能性があります。ただし、非劣性マージンを厳しくすれば、回避できる問題であると考えられています。
プラセボ対照試験が困難な場合の工夫
新薬候補の有効性の検証のためには、原則としてプラセボ対照試験が必要です。特に、自覚症状等プラセボ効果が大きいと想定される場合、疾患の自然経過により症状が変動する場合などではプラセボ対照試験が必要であるとされています。その一方で、倫理上の問題からプラセボ使用が困難となる場合があることは、「プラセボ対照試験の倫理上の問題」の項で述べたとおりです。
プラセボ対照試験が困難な場合の⼯夫の一つの手段として、試験デザインによる対応が考えられます。
ICHの「臨床試験における対照群の選択とそれに関する諸問題(ICH-E10)では、プラセボ対照試験のデザインを修正した方法として以下のものが解説されています。このような修正により、被験者の被るリスクが十分に小さくなる場合には、対照群に標準治療薬ではなくプラセボ投与を行うことが許容される場合があります。
3群⽐較試験 |
2群比較試験(新薬候補の実薬対照試験)にプラセボ対照を追加した試験。 |
上乗せ試験 |
標準治療薬に新薬候補またはプラセボを追加する試験方法。既に受けている標準治療薬が十分には有効でない場合にのみ当該試験デザインが有益とされている。 |
置き換え試験 |
被験者に標準治療薬を有効用量で用いた上で、新薬候補またはプラセボをランダム化により上乗せした後、標準治療薬を少しずつ減量する試験方法。 |
早期離脱・救済治療 |
臨床症状が悪化したり、事前に決めたレベルまで改善しない被験者、対象となる治療で防ごうとした事象が生じた被験者、または他のレスキュー治療が必要となった被験者を早急に試験から離脱させる試験方法。治療を変更する必要が生じたことをエンドポイントとして評価する。 |
短期のプラセボ対照 |
長期のプラセボ治療が受け入れられない状況において、実薬対照試験の開始時点に短期間プラセボ群を用いる試験方法。 |
ランダム化治療中⽌ |
一定期間、新薬候補による治療を受けた被験者が、被験薬治療の継続、またはプラセボ(実薬治療の中止)のいずれかにランダムに割り付けられる試験方法。 |
それぞれの試験デザインには、必ずしも被験者の被るリスクや負担の最小化という観点からは有用でない場合もあります。そのため、修正した試験デザインの実施に当たっては、個別の試験計画に応じた慎重な検討が必要です。また、疾患領域ごとの臨床評価ガイドラインでは、試験デザインの選択とともにプラセボ対照試験の実施に際しての留意点が記載されている場合があります。
また、プラセボ対照試験を実施するにあたっては、治験審査委員会等で十分議論したうえで、科学的に評価するために、プラセボ対照試験を用いることが必要であることについて、患者の理解を得るよう努めることも重要であると考えられます。
シミックヘルスケア・インスティテュート株式会社(CHI) は、SMO(Site Management Organization:治験施設支援機関)として、全国4,000以上の医療機関と提携し、20年以上にわたり各医療機関を支援してまいりました。
これまでの知見・経験を活かし、治験・臨床研究の実施から事務的業務、IRB(治験審査委員会)事務局業務まで、医療機関における治験実施をフルサポートいたします。
まずはお気軽にお問い合わせください。
■関連資料
→SSIカンパニー・SMO事業|シミックヘルスケア・インスティテュート(CHI)










